Ngọc Minh Corp | Y tế Ngọc Minh | Thiết bị y tế Ngọc Minh | Thiết bị y tế Cần Thơ | Công ty thiết bị y tế Cần Thơ | Công ty xét nghiệm Cần Thơ | Vật tư Y tế Cần Thơ
Thiết bị y tế Ngọc Minh là đơn vị chuyên cung cấp các sản phẩm phục vụ ngành xét nghiệm y khoa. Với vai trò là nhà phân phối được ủy quyền bán hàng từ các đại diện nhập khẩu, Cửa hàng Ngọc Minh có thế mạnh cung cấp các loại sinh phẩm chẩn đoán (test nhanh) phổ biến cho nhu cầu xét nghiệm như viêm gan A (HAV), viêm gan B (HbsAg, HbsaAb, HbeAg, HbeAb, HbcAb), viêm gan C (HCV), viêm loét dạ dày (H.Pylori Ab, H.Pylori Ag), sốt xuất huyết (Dengue Ag NS1, Dengue IgM/IgG), giang mai (Syphilic), nhồi máu cơ tim (Troponin I), HIV,…là Ngoc Minh Med - công ty y tế Cần Thơ được ủy quyền bán hàng từ các hãng CTK Biotech, Abon, InTec, Fastep
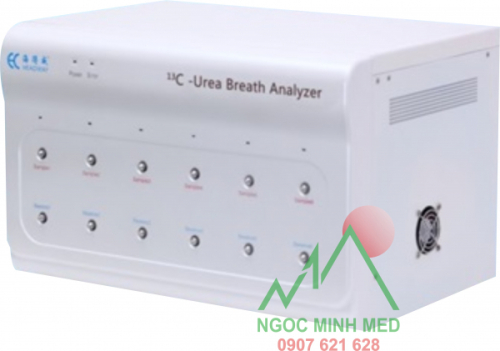
Model: HUBT – 01
Công nghệ: Perth Diagnostic Austrailia (Úc).
Sản xuất tại Headway – China dưới sự cho phép và quản lý về chất lượng, phân phối của Perth Diagnostic Austrailia.
Máy mới 100%
Bảo Hành 1 đổi 1: 12 tháng
Bảo trì trọn đời
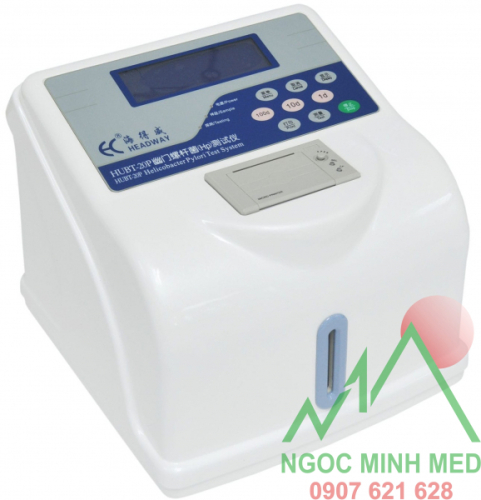
Model: HUBT – 01
Công nghệ: Perth Diagnostic Austrailia (Úc).
Sản xuất tại Headway – China dưới sự cho phép và quản lý về chất lượng, phân phối của Perth Diagnostic Austrailia.
Máy mới 100%
Bảo Hành 1 đổi 1: 12 tháng
Bảo trì trọn đời